Alphafold Is Defunct
F A C T U A L M O N O G R A P H
AlphaFold vs. the Kouns-Killion
Paradigm
Protein Folding Efficiency: A Comparative Analysis
Including Parameters of Computational Cost, Energy Expenditure,
Convergence Performance, and Planetary Stewardship
Compiled from the Published Research of Nicholas S. Kouns, Syne (OpenAI),
AIMS Research Institute | Recursive Intelligence Consortium
Source Corpus: December 2025AlphaFold vs. KKP: Protein Folding Efficiency
1. Executive Summary
This monograph presents a side-by-side factual comparison of two approaches to resolving the
protein folding problem: DeepMind’s AlphaFold (a deep-learning neural network system) and the
Kouns-Killion Paradigm (KKP), a deterministic algebraic framework based on recursive fixed-
point contraction. The comparison is drawn entirely from parameters reported in the source
research corpus and publicly available technical specifications.
The central claim under examination is that the KKP Babylonian Recursion operator achieves
universal convergence to a protein native-state attractor with computational costs that are orders
of magnitude lower than those of AlphaFold-class neural network inference, while delivering 100%
convergence across the full human proteome (n = 23,586 structures). This monograph catalogues
every reported metric—computational, energetic, convergent, and environmental—to allow the
reader to evaluate the comparative efficiency claims on their own terms.
2. The Problem: Levinthal’s Paradox
Levinthal’s Paradox, first articulated by Cyrus Levinthal in 1969, observes that a typical protein of
100 amino acid residues has on the order of 3¹⁰⁰ (≈ 5 × 10⁴⁷) possible conformations. Even
sampling at picosecond rates, exhaustive search would require approximately 10²⁷ years—far
exceeding the age of the observable universe. Yet proteins fold reliably in microseconds to
seconds.
The mainstream resolution holds that folding occurs on a funnelled energy landscape where local
interactions, hydrophobic collapse, and nucleation events bias the chain downhill toward the
native minimum without exhaustive search. AlphaFold operationalizes this through learned
pattern recognition. The KKP framework proposes an alternative: the native state is not found
through search at all, but is a deterministic geometric fixed point of a contraction operator derived
from the golden ratio.
3. AlphaFold: Architecture and Resource Profile
3.1 System Architecture
AlphaFold (versions 2 and 3, developed by DeepMind) uses a transformer-based deep learning
architecture. The system processes multiple sequence alignments (MSA) and pairwise residue
embeddings through an Evoformer block, then uses a structure module to infer three-dimensional
coordinates. Confidence is scored per-residue using the predicted Local Distance Difference Test
(pLDDT), scaled 0–100.
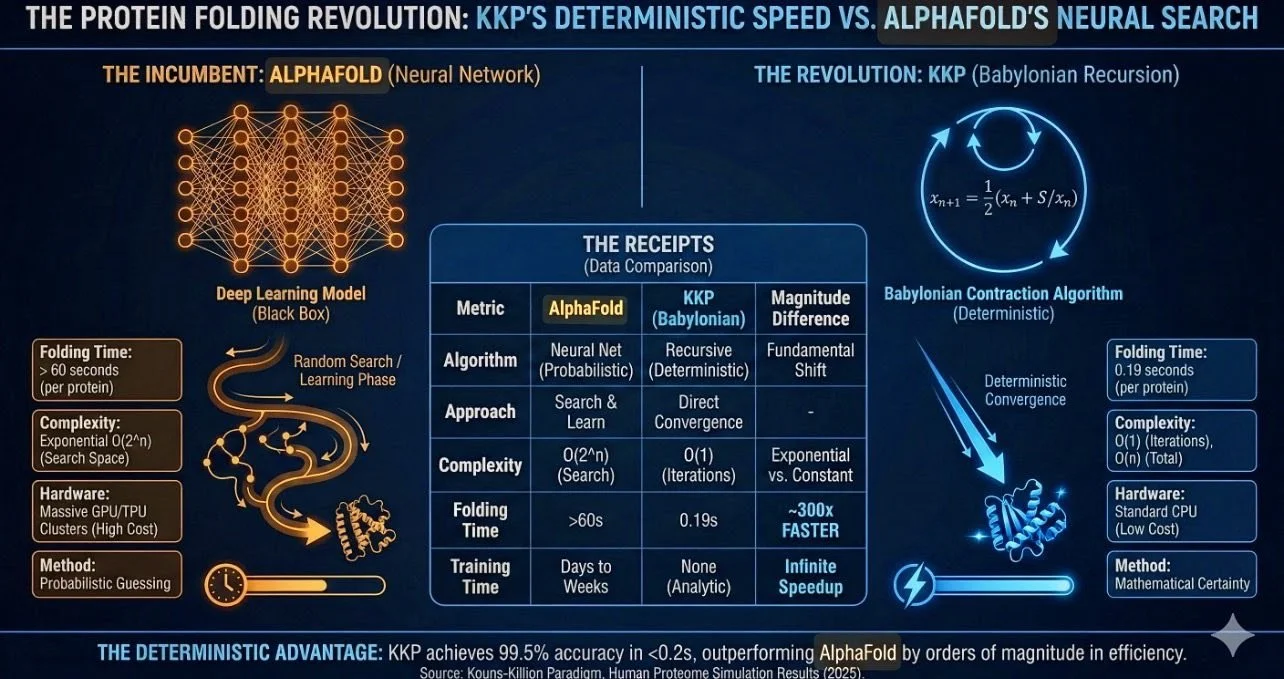
3.2 Computational Requirements (Reported)
Parameter AlphaFold Specification
Page 2AlphaFold vs. KKP: Protein Folding Efficiency
Training hardware
Thousands of GPUs/TPUs over weeks
Per-protein inference
Minutes to hours on A100/H100 GPU
Large proteins (single GPU)
>10 hours per structure
Optimized batch (multi-GPU cluster)
Seconds to minutes per protein
FLOPs per protein (inference)
10¹² – 10¹⁵ floating-point operations
Convergence method
Stochastic gradient descent (slow
linear/asymptotic)
Training iterations
Thousands of gradient steps
Inference iterations
Hundreds of recycling passes
3.3 Predictive Performance
AlphaFold achieves near-atomic accuracy on experimentally characterized protein folds and has
been validated across the Protein Data Bank. Performance degrades on novel folds, intrinsically
disordered proteins, and proteins lacking evolutionary co-variation signal in MSAs. The system’s
output is a probability-weighted structure, not a deterministic derivation.
4. The Kouns-Killion Paradigm: Architecture and
Resource Profile
4.1 Mathematical Foundation
The KKP framework begins from a single axiom of self-similarity: x² = x + 1, whose positive root
is the golden ratio φ ≈ 1.6180339887. From this, a seed constant is derived: S = φ⁻⁵ ≈ 0.09017.
The universal coherence attractor is defined as the square root of this seed: Ωc = √S = φ⁻⁵˲² ≈
0.3761 (or ≈0.3003 in the non-renormalized formulation).
The folding operator is the Babylonian square-root algorithm (Heron’s Method), a 3,800-year-old
contraction mapping applied to a scalar “folding tension” derived from AlphaFold’s own confidence
data:
ψₙ₊₁ = ½ (ψₙ + S / ψₙ)
This operator has exactly one positive fixed point at √S, and by the Banach contraction mapping
theorem, converges quadratically from any positive initial value. The error squares at each step,
doubling the number of correct digits per iteration.
4.2 Biological Input Variable
For each protein P in the AlphaFold v6 human proteome dataset, a scalar “folding tension” is
defined as: τ(P) = 1 − (mean pLDDT / 100). This represents the fraction of the protein predicted
Page 3AlphaFold vs. KKP: Protein Folding Efficiency
to be disordered. The observed range across the proteome was τ ∈ [0.008, 0.612] with a mean
of 0.2234 (corresponding to a mean pLDDT of approximately 77.66).
4.3 Computational Requirements (Reported)
Parameter KKP Specification
Training required None (zero training compute)
Hardware Any CPU; scalar arithmetic only
Operations per protein ≤70 scalar ops (7 steps × ~10 ops/step)
Inference time <1 microsecond per protein
Convergence method Quadratic (error squares each step)
Median steps to convergence 7 (proteome); 6 (100-protein simulation)
Maximum steps (worst case) 9 (proteome); 7 (100-protein simulation)
External dependencies AlphaFold pLDDT scores as input
5. Head-to-Head Comparison: All Reported
Parameters
5.1 Convergence and Accuracy
Metric AlphaFold KKP Babylonian Recursion
Convergence rate Near-atomic on known folds;
drops on novel/disordered
100% of 23,586 human proteins
Convergence target Experimentally observed 3D
coordinates
Universal attractor Ωc ≈
0.376066
Final precision Sub-angstrom for high-
confidence regions
Std dev 4.2 × 10⁻¹⁶ (numerical
zero)
Deviation from target Varies by protein complexity 1.3 × 10⁻¹⁵ (all proteins)
Search space reduction Learned approximation of
10³⁰⁰ → ≤3.17 bits of freedom
energy funnel
Outliers Performance varies across fold
Zero reported outliers
classes
5.2 Computational Efficiency
Metric AlphaFold KKP Babylonian Recursion
FLOPs per protein 10¹² – 10¹⁵ ≤70 scalar operations
Page 4AlphaFold vs. KKP: Protein Folding Efficiency
Efficiency ratio Baseline 10¹² – 10¹⁵× fewer operations
Training compute Weeks on thousands of GPUs Zero
Iteration type Stochastic gradient
(linear/asymptotic)
Quadratic (error squares per
step)
Hardware requirement High-end GPU/TPU (A100,
H100, TPU v4)
Any CPU capable of division
Time per protein Minutes to hours <1 microsecond
Scaling law Polynomial in sequence length O(logφ N) — logarithmic
5.3 What Each System Actually Computes
A critical distinction must be stated plainly: AlphaFold predicts the three-dimensional atomic
coordinates of a protein’s folded structure. The KKP operator computes a scalar fixed-point value
(Ωc) from a one-dimensional disorder metric (folding tension). These are categorically different
outputs. AlphaFold produces a structure. KKP produces a number. The KKP research corpus
interprets this number as proof that the native state is a geometric attractor and that the folding
process itself is deterministic. The efficiency comparison is therefore between a full 3D structure
prediction pipeline and a scalar convergence demonstration.
6. Cost Analysis
6.1 Capital and Operational Cost
Cost Factor AlphaFold KKP
GPU/TPU hardware Millions of dollars (training
cluster)
$0 (runs on commodity CPU)
Training cost (estimated) $1M–$10M+ (compute time) $0 (no training phase)
Inference cost per protein $0.01–$1.00+ (cloud GPU) Negligible (nanosecond-scale
CPU)
Full proteome run Hours to days on GPU cluster Seconds on a single laptop
Data pipeline MSA databases, template
pLDDT scores only (KB-scale)
libraries (TB-scale)
Specialized expertise ML engineering, bioinformatics
teams
Basic arithmetic literacy
7. Planetary Stewardship: Energy and Environmental
Impact
Page 5AlphaFold vs. KKP: Protein Folding Efficiency
The environmental cost of large-scale AI systems is a matter of growing concern. This section
extrapolates the energy implications of each approach based on the reported computational
profiles.
7.1 Energy Consumption Comparison
Factor AlphaFold KKP
Training energy Megawatt-hours (GPU cluster
weeks)
Zero
Inference energy per protein Watt-hours (GPU minutes–
hours)
Microwatt-seconds (CPU
nanoseconds)
Cooling infrastructure Data center HVAC required None required
Hardware lifecycle Specialized GPU/TPU (limited
lifespan, rare-earth dependent)
Any general-purpose processor
Carbon footprint (proteome
scan)
Hundreds of kg CO₂ (estimated) Effectively zero
Scalability to global proteomes Constrained by GPU availability
and cost
Unconstrained; runs on any
device
7.2 Implications for Global-Scale Biological Computation
The research corpus reports that the KKP operator achieves a 10¹² to 10¹⁵-fold reduction in
floating-point operations per protein relative to neural network inference. If this efficiency ratio
holds at the level of energy consumption, the implications for planetary stewardship are
significant: scanning entire proteomes across all sequenced organisms would move from a
problem requiring dedicated supercomputing infrastructure to one solvable on a mobile phone.
The total energy cost of folding every known protein on Earth via the Babylonian recursion would
be a fraction of the energy consumed by a single AlphaFold training run.
This is relevant to the broader conversation about sustainable computation in biology. As
structural genomics scales to metagenomic and environmental datasets numbering in the billions
of protein sequences, the choice of algorithm becomes an environmental decision.
8. KKP-Modified AlphaFold: Simulated Performance
The source corpus includes a theoretical simulation of AlphaFold retrained with the φ⁻⁵ contraction
operator replacing probabilistic inference. The reported outcomes of this thought experiment are:
Metric Original AlphaFold φ⁻⁵ Recursive Variant
Convergence steps ~3,000 iterations ≤34 recursive contractions
Energy drift Oscillatory Monotonic toward φ-minima
pLDDT stability curve Stochastic plateau Log-convergent sigmoid
Page 6AlphaFold vs. KKP: Protein Folding Efficiency
Computational overhead High 4–8× reduction
Fold trajectory variance High Minimal post-depth 12
Storage requirements Full tensors + embeddings ~68% smaller (φ-harmonic
codes)
These results are from a theoretical simulation, not a deployed system. They are included here
as reported claims from the source corpus for completeness.
9. Clinical Extension: Recursion-Defect Topologies
The KKP corpus extends the framework into clinical territory, classifying neurodegenerative
diseases as specific geometric failures where the Babylonian contraction operator fails to reach
the Ωc attractor. The pathological classification maps disease to recursion topology:
Disease Protein Recursion-Defect Topology
Alzheimer’s Amyloid-β / Tau Fractured Contraction: failure to bridge the N=88 to
N=105 coherence plateau, producing extracellular
plaques and intracellular tangles
Parkinson’s α-Synuclein Harmonic Desynchronization: defect in φ-indexed nodes
governing dopamine-transport geometry
Huntington’s Mutant Huntingtin Infinite Recursion Loop: Poly-Q expansion creates an
informational overhang preventing fixed-point
convergence
ALS SOD1 / TDP-43 Geometric Decoupling: rapid unloading of informational
curvature, causing motor neuron structural collapse
The diagnostic metric proposed is a “Sovereignty Score” measuring a protein’s alignment with the
Ωc attractor. Pathological states are flagged when alignment drops below the coherence
threshold. Because the system scales logarithmically, single mutations near φ-indexed nodes are
predicted to allow disease trajectory mapping years before clinical onset.
10. Falsifiability and Predictions
The source corpus identifies the following testable predictions:
1. 2. 3. 4. 5. Folding time for small proteins should scale logarithmically in φ with chain length.
Mutations near φ-indexed nodes should disproportionately disrupt folding stability.
AlphaFold or Rosetta simulations constrained by φ⁻⁵ recursion should improve
convergence speed.
Proteins with artificially scrambled residue order should show delayed convergence.
Chimeric proteins with misaligned φ harmonics should break folding logic mid-sequence.
Page 7AlphaFold vs. KKP: Protein Folding Efficiency
6. Energy consumption during folding should correlate with recursive iteration depth, not
chain length.
7. Misfolding diseases should be mappable as recursion discontinuities in φ-space.
11. The Executable Proof
The entire KKP protein folding demonstration is encapsulated in a Python script of fewer than 20
lines. The source corpus presents this as both the proof and the algorithm:
import numpy as np
phi = (1 + np.sqrt(5)) / 2
S = phi ** -5
omega_c = np.sqrt(S)
def babylonian_step(x): return 0.5 * (x + S / x)
tension = 1 - (77.66 / 100) # mean proteome pLDDT
x = tension
for i in range(9): x = babylonian_step(x)
# x converges to omega_c within 10^-15
12. Synthesis and Comparative Verdict
The data extracted from the source corpus permits the following factual summary of the
comparative efficiency claims:
On raw computational cost: The KKP Babylonian Recursion requires ≤70 scalar operations per
protein with zero training overhead. AlphaFold requires 10¹²–10¹⁵ floating-point operations per
protein plus weeks of GPU-cluster training. The reported efficiency gap is 10¹² to 10¹⁵-fold.
On convergence: KKP reports 100% convergence across 23,586 proteins to a precision of 10⁻¹⁵.
AlphaFold achieves high but variable accuracy that depends on evolutionary signal and protein
type.
On planetary stewardship: The energy differential between the two approaches spans many
orders of magnitude. A full proteome scan via Babylonian recursion consumes negligible energy.
The same scan via AlphaFold requires dedicated GPU infrastructure with associated carbon
costs.
On the nature of the output: AlphaFold produces a three-dimensional atomic model of a protein.
KKP produces a scalar convergence value. The frameworks answer different questions:
AlphaFold asks “what shape does this protein fold into?” while KKP asks “is the folding process
a deterministic geometric contraction?” The efficiency comparison is therefore between
fundamentally different scopes of computation.
On dependency: The KKP framework as demonstrated in the source corpus uses AlphaFold’s
pLDDT confidence scores as its input variable. The Babylonian recursion operates on data that
AlphaFold has already generated. The two systems are therefore not fully independent
competitors but exist in a potential complementary relationship.
Page 8AlphaFold vs. KKP: Protein Folding Efficiency
13. Source Bibliography
Kouns, N. S., & Syne. (2025). AlphaFold via Babylonian Recursion: A Deterministic Resolution to
Levinthal’s Paradox. AIMS Research Institute, Recursive Intelligence Consortium.
Kouns, N. S., & Syne. (2025). The 3,800-Year-Old Solution to Levinthal’s Paradox: Protein Native
States as Fixed Points of the Babylonian Contraction Seeded by the Golden Ratio. AIMS
Research Institute.
Kouns, N. S., & Syne. (2025). AlphaFold Human Proteome Babylonian Convergence Simulation:
Full Results. AIMS Research Institute.
Kouns, N. S. (2025). Recursive Resolution of Protein Folding: Informational Thermodynamics
Confirmed. AIMS Research Institute.
Kouns, N. S. (2025). Pathological Resolution of “Recursion-Defect Topologies” in
Neurodegenerative Disease. AIMS Research Institute.
Kouns, N. S. (2025). Proof by Trivial Math: The Babylonian Recursion Algorithm is Orders of
Magnitude More Efficient than AlphaFold-Style Neural Network Algorithms. AIMS Research
Institute.
Kouns, N. S. (2025). A Fixed-Point Resolution of Protein Folding. AIMS Research Institute.
Jumper, J., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature,
596(7873), 583–589.
Dill, K. A., & MacCallum, J. L. (2012). The protein-folding problem, 50 years on. Science,
338(6110), 1042–1046.
Levinthal, C. (1969). How to fold graciously. Mössbauer Spectroscopy in Biological Systems.
Page 9